
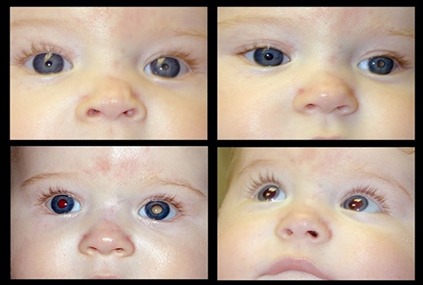
Retinoblastoma is a cancer of the eye that is usually diagnosed by the age of 6 years, with the majority of these cases seen by age 3 year. The disease can be seen at birth, and it can be unilateral or bilateral. Diagnosis of retinoblastoma is made earlier in bilateral disease. Here we can see a typical unilateral presentation where the pupillary reflex in the affected eye shines white. In bilateral disease the eyes are usually affected to a different extent. Unilateral cases are often non-hereditary. Bilateral disease is always hereditary. Since unilateral cases are often discovered much later, it is often necessary to enucleate the eye because vision in the affected eye is not salvageable. Bilateral cases are often discovered much earlier and treated with a combination of enucleation of one eye in some cases, and chemotherapy, radiation, transpupillary thermal treatment (TTT), and cryotherapy.
- Cancer of the Eye
- Diagnosis: Birth-~6 years
- Unilateral or Bilateral
- ~3% of Pediatric Cancers.
Statistics of Retinoblastoma: The most important thing to learn from these statistics is that bilateral disease is genetic, meaning that each offspring has a 50% chance of having the disease. Unilateral retinoblastoma is most likely a somatic mutation occurring in the eye and is not hereditary. However it must be understood that 15% of unilateral cases can be hereditary. Genetic testing is needed to determine which is which Statistics of Retinoblastoma Complete.
- 85% sporadic
- 15% familial
- 60% unilateral
- 40% bilateral
- 85% of unilateral have somatic mutations, 15% have germinal
- 90% of bilateral have germinal mutations, 10% somatic
- Trilateral retinoblastoma affecting also the pineal is extremely rare
![]() Epidemiology: The estimated incidence of retinoblastoma is 1 in 16,000-18,000 births annually with between 7000 and 8000 new cases per year worldwide. The annual incidence is 3.5 per million children younger than 15 and 11.8 per million children younger than 5 years of age. While survival rates in the United States are nearly 100%, they are much lower in developing nations. Survival rates range from 80-89% in more developed Latin American countries to as low as 20-46% in certain African countries. More than 90% of children with retinoblastoma live in low and middle-income countries (LMIC), but those countries have 90% of the cases presenting with metastatic disease, and virtually all the cases that abandon therapy.
Epidemiology: The estimated incidence of retinoblastoma is 1 in 16,000-18,000 births annually with between 7000 and 8000 new cases per year worldwide. The annual incidence is 3.5 per million children younger than 15 and 11.8 per million children younger than 5 years of age. While survival rates in the United States are nearly 100%, they are much lower in developing nations. Survival rates range from 80-89% in more developed Latin American countries to as low as 20-46% in certain African countries. More than 90% of children with retinoblastoma live in low and middle-income countries (LMIC), but those countries have 90% of the cases presenting with metastatic disease, and virtually all the cases that abandon therapy.
As a result of lower survival rates, there are an estimated 3000 to 4000 deaths annually due to retinoblastoma. The discrepancies in survival rates emphasize the potential to reduce retinoblastoma-caused through timely diagnosis and proper treatment.
Retinoblastoma Cell-of-Origin: Definitive determination of the cell-of-origin and understanding how tumor cells exploit the mechanisms of such susceptible cells for malignant transformation may facilitate development oftreatments that specifically target tumor cells, while sparing normal retinal tissue. In this section, we discuss direct and circumstantial evidence supporting a cone cell-of-origin and elucidate why cone cells are susceptible to cancerous transformation. Cone Cells Proliferate in Response to Retinoblastoma Protein (prb) Loss, While Cone-Enriched Genes are Prominently Expressed in Retinoblastoma.
Genetics of retinoblastoma: Out of the newly diagnosed cases of retinoblastoma only 6% are familial while 94% are sporadic. Bilateral retinoblastomas involve germinal mutations in all cases. Approximately 15% of unilateral sporadic retinoblastoma is caused by germinal mutations affecting only one eye while the 85% are sporadic Genetic counseling is an important aspect in the management of retinoblastoma. In patients with a positive family history, 40% of the siblings would be at risk of developing retinoblastoma and 40% of the offspring of the affected patient may develop retinoblastoma. In patients with no family history of retinoblastoma, if the affected child has unilateral retinoblastoma, 1% of the siblings are at risk and 8% of the offspring may develop retinoblastoma. In cases of bilateral retinoblastoma with no positive family history, 6% of the siblings and 40% of the offspring have a chance of developing retinoblastoma.
Clinical Manifestations of Retinoblastoma: Leucocoria is the most common presenting feature of retinoblastoma, followed by strabismus, painful blind eye and loss of vision. Table 1 lists the common presenting features of retinoblastoma.
Table 1. Common presenting features of retinoblastoma
1. Leucocoria 56%
2. Strabismus 20%
3. Red painful eye 7%
4. Poor vision 5%
5. Asymptomatic 3%
6. Orbital cellulitis 3%
7. Unilateral mydriasis 2%
8. Heterochromia iridis 1%
9. Hyphema 1%
The clinical presentation of retinoblastoma depends on the stage of the disease.
• Early lesions are likely to be missed, unless an indirect ophthalmoscopy is performed. The tumor appears as a translucent or white fluffy retinal mass . The child may present with strabismus if the tumor involves the macula, or with reduced visual acuity. Moderately advanced lesions usually present with leucocoria due to the reflection of light by the white mass in the fundus . As the tumor grows further, three patterns are usually seen.
• Endophytic, in which the tumor grows into the vitreous cavity . A yellow white mass progressively fills the entire vitreous cavity and vitreous seeds occur. The retinal vessels are not seen on the tumor surface.
• Diffuse infiltrating tumor, in which the tumor diffusely involves the retina causing just a placoid thickness of the retina and not a mass. This is generally seen in older children and usually there is a delay in the diagnosis. Advanced tumors manifest with proptosis secondary to optic nerve extension or orbital extension and systemic metastasis. Retinoblastoma can spread through the optic nerve with relative ease especially once the lamina cribrosa is breached. Orbital extension may present with proptosis and is most likely to occur at the site of the scleral emissary veins. Systemic metastasis occurs to the brain, skull, distant bones and the lymph nodes. Some of the atypical manifestations of retinoblastoma include pseudohypopyon, spontaneous hyphema, vitreous hemorrhage, phthisis bulbi and preseptal or orbital cellulitis.
Diagnosis of retinoblastoma: A thorough clinical evaluation with careful attention to details, aided by ultrasonography B-scan helps in the diagnosis.
Computed tomography and magnetic resonance imaging are
generally reserved for cases with atypical manifestations and diagnostic dilemma and where extraocular or intracranial tumor extension is suspected.
A child with suspected retinoblastoma necessarily needs complete ophthalmic evaluation including a dilated fundus examination under anaesthesia. The intraocular pressure is measured and the anterior segment is examined for neovascularization, pseudohypopyon, hyphema, and signs of inflammation. Bilateral fundus examination with 360 degree scleral depression is mandatory. Direct visualization of the tumor by an indirect ophthalmoscope is diagnostic of retinoblastoma in over 90% of cases. RetCam is a wide-angle fundus camera, useful in accurately documenting retinoblastoma and monitoring response to therapy. Ultrasonography B-scan shows a rounded or irregular intraocular mass with high internal reflectivity representing typical intralesional calcification. Computed tomography delineates extraocular extension and can detect an associated pinealoblastoma. Magnetic resonance imaging is specifically indicated if optic nerve invasion or intracranial extension is suspected. On fluorescein angiography, smaller retinoblastoma shows minimally dilated feeding vessels in the arterial phase, blotchy hyperfluorescence in the venous phase and late staining.
Management of Retinoblastoma: The primary goal of management of retinoblastoma is to save life. Salvage of the organ (eye) and function (vision) are the secondary and tertiary goals respectively. The management of retinoblastoma needs a multidisciplinary team approach including an ocular oncologist, pediatric oncologist, radiation oncologist, radiation physicist, and an ophthalmic oncopathologist. The management strategy depends on the stage of the disease – intraocular retinoblastoma, retinoblastoma with high-risk characteristics, orbital retinoblastoma and metastatic retinoblastoma. Through clinical evaluation with careful attention to details, aided by ultrasonography B-scan helps in the diagnosis. Computed tomography and magnetic resonance imaging are generally reserved for cases with atypical manifestations and diagnostic dilemma and where extraocular or intracranial tumor extension is suspected. A child with suspected retinoblastoma necessarily needs complete ophthalmic evaluation including a dilated fundus examination under anaesthesia.The intraocular pressure is measured and the anterior segment is examined for neovascularization, pseudohypopyon, hyphema, and signs of inflammation. Bilateral fundus examination with 360 degree scleral depression is mandatory. Direct visualization of the tumor by an indirect ophthalmoscope is diagnostic of retinoblastoma in over 90% of cases.RetCam is a wide-angle fundus camera, useful in accurately documenting retinoblastoma and monitoring response to therapy . Ultrasonography B-scan shows a rounded or irregular intraocular mass with high internal reflectivity representing typical intralesional calcification. Computed tomography delineates extraocular extension and can detect an associated pinealoblastoma.Magnetic resonance imaging is specifically indicated if optic nerve invasion or intracranial extension is suspected.On fluorescein angiography, smaller retinoblastoma shows minimally dilated feeding vessels in the arterial phase, blotchy hyperfluorescence in the venous phase and late staining.
Examination: Anything causing a white pupil reflex as the only sign suggesting the presence of retinoblastoma should lead the examiner to consider the following:
- The patient is usually young, under age 2 years.
- The retina must be involved.
- Anterior globe involvement is always secondary to primary retinal involvement.
- This is usually the only problem with the child except in the rare case of the 13q deletion, where children have multiple systemic problems.
- It is always possible that this is a photographic artifact.
- Ultrasound
- The findings with B-scan ultrasound are typical and are shown here.
- Normal axial length
- Solid mass
- Highly reflective
- Shadowing
- Re-duplication
- Computed Tomography
- Intraocular mass
- Calcifications
- Heterogeneous
- High density
- Enhancement of soft tissue mass
Considerations:
- Recent studies suggest possible radiation-induced tumors secondary to CT scanning.
- MRI has replaced CT in most cases where MRI provides sufficient information.
- Seeing calcification in a tumor like this confirms the diagnosis of retinoblastoma
- complete retina exam with dilated pupil using indirect ophthalmoscope) and then:
- Ultrasonography
Neuroimaging: CT and MRI
Thorough general physical examination
Followed by (if indicated):
- DNA testing in peripheral and in tumor cells
- Spinal tap
- Bone marrow aspirate/biopsy
- Skeletal survey
The diagnosis can usually be made early on. Further examination is required for staging and special laboratory testing is required to determine the genetics.
Retina examination with the indirect ophthalmoscope This view of the retina obtained with a RetCam gives a view of more of the retina than can be seen with the indirect ophthalmoscope, but these are two types of retinoblastoma that are typical. Ultrasound The findings with B-scan ultrasound are typical and are shown here.
- Normal axial length
- Solid mass
- Highly reflective
- Shadowing
- Re-duplication
- CT Scan vs. MRI
In the example shown here, the MRI fails to detect the tumor while the CT scan shows the tumor with calcium in studies done on the same patient.
Classification:
Group 1: Very favorable
A. solitary tumor < 4 DD at or behind equator
B. multiple tumors, not over 4 DD, all at or behind
Group 2: Favorable
A. Solitary tumor, 4 – 10 DD at or behind equator
B. Multiple tumors, 4 – 10 DD behind equator
Group 3: Doubtful
A. Any tumor anterior to equator
B. Solitary tumor, > 10 DD, behind equator
Group 4: Unfavorable
A. Multiple tumors, some > 10 DD
B. Any lesion extending anterior to ora serrata
Group 5: Very unfavorable
A. Massive tumors involving > 1/2 of retina
B. Vitreous seeding
Treatment: Retinoblastoma treatment primarily aims to save the patient’s life and then their globe and vision. Although retinoblastoma is the most curable cancer with a high survival rate, advanced retinoblastoma limits globe and vision salvaging and risks the patient’s life because of metastasis. Next, we summarize the classification and staging systems and high-risk histopathological features of retinoblastoma, which are crucial for disease management and prognosis. Additionally, we discuss the current treatments, correlations between pathological risks and clinical features, which assist in guiding treatment and may facilitate globe preservation, and development of targeted therapeutics based on the biological mechanisms underlying the disease.
1. Classification and Staging Systems The classification schemes have been developed to predict success of globe survival in intraocular retinoblastoma based on treatment modalities. In the external beam radiotherapy (EBRT) era, Reese and Ellsworth classification was widely used. In the systemic chemotherapy era, the International Intraocular Retinoblastoma Classification (IIRC) was developed by Murphree [109] and modified into a scheme known as the Intraocular Classification of Retinoblastoma (ICRB) which is found to successfully predict the outcome of intravenous chemotherapy (IVC) . These systems classify tumors, according to their size, location, and additional features including tumor seeds into groups A–E from very low to very high risk. However, significant discrepancies remain for group assignments using IIRC and ICRB, especially group D and E eyes, leading to inconsistency in clinical research reports and making comparisons difficult between treatment centers. The International Retinoblastoma Staging System (IRSS) and Tumor, Node, Metastasis (TNM) staging system have been used to cover the whole spectrum of retinoblastoma. Recently, the American Joint Committee on Cancer (AJCC) developed the 8th version of AJCC retinoblastoma staging. This new version has acknowledged the importance of detecting the heritable trait (H) in addition to TNM and is proposed to predict globe salvage, metastasis and patient survival more accurately.
2. High-Risk Histopathological Features Risk of tumor dissemination increases as growing tumors invade into the optic nerve and highly vascularized choroidal tissue. Tumor cells may spread into the cranial cavity, bone, bone marrow, or lymph nodes. An important histological feature examined in enucleated globes and considered as a high-risk factor for distal metastasis is retrolaminar optic nerve invasion including the tumor at the surgical margin and massive choroidal invasion at ≥3 mm in diameter. Tumor invasion into the anterior segment and any degree of concomitant non-massive choroidal and prelaminar/laminar optic nerve invasions are considered as prognostic risk factors. These high-risk histopathological features are indications for adjuvant chemotherapy at most clinics. Current Treatments Treatment of retinoblastoma is challenging and has been markedly developed in recent decades. In the 1960s, EBRT was extensively used for the treatment of intraocular retinoblastoma. However, with the advent of systemic chemotherapy, the popularity of EBRT has declined because of its side effects such as development of secondary malignancies and radiation-induced complications.
Current treatments: Treatment of retinoblastoma is challenging and has been markedly developed in recent decades. In the 1960s, EBRT was extensively used for the treatment of intraocular retinoblastoma. However, with the advent of systemic chemotherapy, the popularity of EBRT has declined because of its side effects such as development of secondary malignancies and radiation-induced complications.
Future Treatments: Cone-associated molecules TRβ1 and TRβ2 are linked with prb pathway at least through controlling SKP2 activity in the pRb-SKP2-p27 axis, while some other molecules, such as MDM2, E2F1, and SYK, also promote tumorigenesis. This knowledge would facilitate designing targeted approaches that are expected to be effective and less toxic.








